ACI Clinical is part of WCGclinical.com
If you were trying to reach aciclinical.com, you’re in the right place!
As part of our ongoing efforts to simplify and improve our client experience, we have consolidated the ACI Clinical website to wcgclinical.com.
This does not represent any changes to our service to you, and there are no actions you or your teams need to take at this time.
Below, you’ll find links to our solutions and important resources. If you have any questions or concerns, please contact us.
Page Navigation:
Explore WCG Solutions

Endpoint Adjudication
Through our decades of focused adjudication experience, we have developed best practices around the Endpoint Adjudication process to lower the risk of variation in important clinical trial outcome events.

Data Monitoring Committee
Our managed Data Monitoring Committees deliberate over un-blinded data to provide recommendations on how trials should proceed. Our streamlined approach ensures patient safety oversight and effective decision-making.

Statistical Consulting
Our statistical consulting services enrich clinical development plans for sponsors who need additional statistical capabilities. This includes protocol and trial design, statistical analysis plan development, and statistical oversight and guidance.
Our expert committees are supported by purpose-built adjudication technology
Our proprietary AIMS® technology is purpose-built for endpoint adjudication committee management. AIMS streamlines processes and diminishes administrative burden, allowing committee members to focus on providing their expert assessments. AIMS makes online data review and communication as seamless as possible for committee members, sponsors and other personnel spanning the globe.
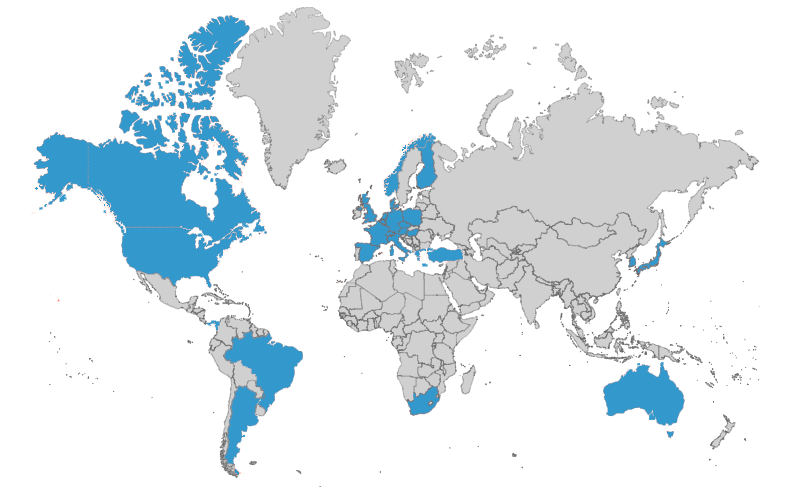
When you need thorough insights, turn to a network of 1,000+ global experts
WCG’s committees leverage our network of 1,000+ medical, statistical, and safety experts who serve as committee members and advisors to clinical trial sponsors.
WCG collaborates with our clients to determine any geographic, scientific expertise, or regulatory needs that would comprise the ideal member profiles for any new project.
Important Resources

Expert Endpoint Adjudication and Data Monitoring Committee Solutions Sell Sheet

The Top 5 Benefits of Outsourcing Expert Committee Management to an Independent Provider

See How WCG Helped a Mid-tier Biopharma Respond to an Urgent FDA Request, and Adjudicate 400 Endpoints in Just a Few Short Weeks
Contact Us
Have a question or want to speak with our sales or support teams?
Use this form to contact us.
Office Information:
401 Plymouth Road, Suite 610
Plymouth Meeting, PA 19462
Phone: +1 (484) 429-7200
Fax: +1 (484) 429-7201